- Letter to the Editor
- Open access
- Published:
PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation
Molecular Cancer volume 16, Article number: 112 (2017)
Abstract
BRAF inhibitors (BRAFi) are standard of care for the treatment of BRAF V600 mutation-driven metastatic melanoma, but can lead to paradoxical activation of the mitogen-activated protein kinase (MAPK) signalling pathway. This can result in the promotion of precancerous lesions and secondary neoplasms, mainly (but not exclusively) associated with pre-existing mutations in RAS genes. We previously reported a patient with synchronous BRAF-mutated metastatic melanoma and BRAF wt/KRAS G12D-metastatic colorectal cancer (CRC), whose CRC relapsed and progressed when treated with the BRAF inhibitor dabrafenib (GSK2118436). We used tissue from the resected CRC metastasis to derive a cell line, LM-COL-1, which directly and reliably mimicked the clinical scenario including paradoxical activation of the MAPK signalling pathway resulting in increased cell proliferation upon dabrafenib treatment. Novel BRAF inhibitors (PLX8394 and PLX7904), dubbed as “paradox breakers”, were developed to inhibit V600 mutated oncogenic BRAF without causing paradoxical MAPK pathway activation. In this study we used our LM-COL-1 model alongside multiple other CRC cell lines with varying mutational backgrounds to demonstrate and confirm that the paradox breaker PLX8394 retains on-target inhibition of mutated BRAF V600 without paradoxically promoting MAPK signalling.
Introduction
A number of studies have demonstrated BRAFi-induced paradoxical activation, particularly when RAS is hyperactivated [1,2,3,4]. This is most commonly manifested as promotion of both benign and malignant hyperproliferative squamous cutaneous lesions in patients treated with BRAFi [5, 6]. Of greater concern is the increased incidence of secondary primary melanomas [7], and the reported emergence of RAS-driven cancers [8,9,10] including our own CRC case study in which combined treatment with dabrafenib and the MEK inhibitor trametinib (GSK 1120212, GlaxoSmithKline) was insufficient to block disease progression [11]. We subsequently established a cell line (LM-COL-1) from the colon cancer metastasis which was able to recapitulate what was observed in the patient during BRAFi treatment. The cell line provided us with a relevant model in which to investigate BRAFi and paradoxical MAPK activation.
In view of the frequency of RAS mutations in CRC [12] and pancreatic cancer [13], and the unknown prevalence of occult MAPK activating mutations in the population at large, it is anticipated that drug-promoted cancers will continue to emerge as a serious clinical problem in patients receiving BRAFi [1]. Consequently, a new generation of BRAFi termed “paradox breakers”, such as PLX8394 and PLX7904 (Plexxikon), has been developed [14,15,16].
Findings
Firstly, we compared the on-target efficacy of PLX8394 (Plexxikon, Berkeley, CA) and the classical BRAFi, vemurafenib, by treating a BRAF V600E melanoma cell line, LM-MEL-64, and a BRAF wt /RAS wt melanoma cell line, LM-MEL-39 with both drugs (Additional file 1: Material and Methods). Strong MAPK pathway inhibition in LM-MEL-64 was demonstrated by an 80.3 ± 2.4% (mean ± SD) reduction of pERK at the 1 μM dose relative to control, while little or no change in pERK was observed in LM-MEL-39 (Additional file 2: Figure S1).
Since paradoxical activation of MAPK signalling appeared to have driven the growth of the colorectal cancer in our CRC case study [11], we examined whether this could be replicated in the LM-COL-1 cell line and additional colorectal cancer cell lines with varying mutational status, and whether this effect could be mitigated by use of PLX8394. The cell lines and their mutational status used in this study are shown in Table 1. Consistent with our previous findings, the BRAFi vemurafenib induced a dose-dependent paradoxical increase in the levels of pMEK and pERK in LM-COL-1 at the 1 μM dose of 72.1 ± 24.5% and 160.2 ± 18.0% (mean ± SD), respectively. In contrast, treatment with the paradox breaker PLX8394 had minimal effect on pMEK and pERK in this cell line (Fig. 1a, c, and e). Similar effects could be seen in the two additional BRAF wt / KRAS G12D colon cancer cell lines, ALA and LS513 (Fig. 1a, c, and e), and were also observed when we applied the same treatments on the BRAF wt / KRAS G13D colon cancer cell line HCT 116 (Additional file 3: Figure S2). Conversely, both vemurafenib and PLX8394 decreased MEK1/2 and ERK1/2 phosphorylation in the BRAF V600E colon cancer cell lines LIM2405 and COLO 201 (Fig. 1b, d, and f).
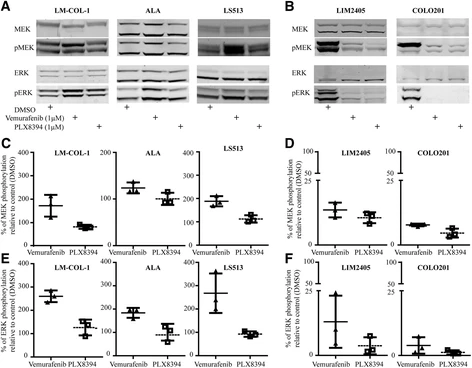
Effect of the BRAF inhibitors vemurafenib and PLX8394 on the MAPK pathway in colorectal cancer cell lines. Cells were treated with DMSO, vemurafenib at 1 μM, or PLX8394 at 1 μM for 6 h. a, b Representative Western blot of a panel of BRAF wt /KRAS G12D (LM-COL-1, ALA, and LS513) and BRAF V600E /KRAS wt (LIM2405 and COLO 201) colorectal cancer cell lines after treatment with DMSO control or BRAF inhibitors. Western blots were probed for total and phosphorylated MEK1/2 and ERK1/2. The blots are representative of three independent experiments. Total ERK served as a loading control. Western blot signal intensity was quantified and used to measure protein level relative to control. c, d Densitometry of MEK1/2 phosphorylation demonstrating paradoxical activation by vemurafenib in KRAS-mutated cell lines and BRAFi sensitivity in BRAF V600E mutated cell lines LIM2405 and COLO 201. e, f Densitometry of ERK1/2 phosphorylation in the same cell lines as shown in c and d. In panels c–f the total protein:phosphorylated ratio is expressed as the mean ± SD of three independent replicates relative to DMSO-treated control
To assess the functional effects of these inhibitors, proliferation assays were performed after 72 h treatment with either vemurafenib or PLX8394 across a range of concentrations. Consistent with the increase in MAPK signalling, proliferation of ALA, LS513, LM-COL-1, and HCT 116 was enhanced when treated with vemurafenib, but not with PLX8394 (Fig. 2a–c, and Additional file 3: Figure S2d). Notably, the largest effect on vemurafenib-induced cell proliferation was observed at the clinically achievable dose of 0.5 μM for ALA and LS513. Western blot inlays from signalling analysis of vemurafenib at concentrations that resulted in the greatest effect of increased proliferation, 0.5 μM for ALA and LS513, 1 μM for LM-COL-1, and 0.1 μM for HCT 116, demonstrate paradoxical increase of pERK in these cell lines (Fig. 2a–c, and Additional file 3: Figure S2a, b and c).
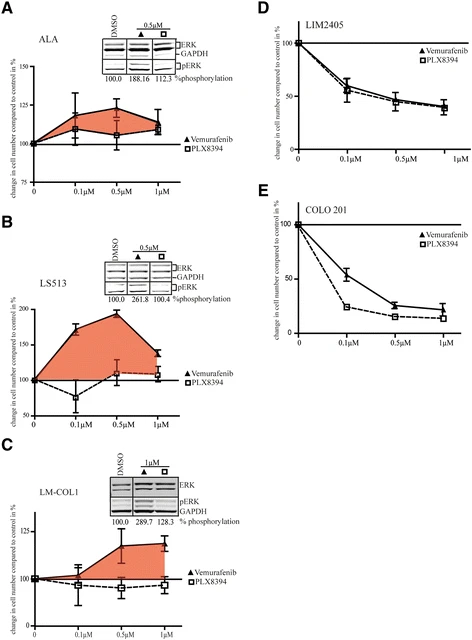
The effect of vemurafenib and PLX8394 on proliferation and survival of BRAF wt / KRAS G12D and BRAF V600E / KRAS wt colorectal cancer cell lines. Inhibitors were used at 0 (DMSO control), 0.1, 0.5, and 1 μM. Cell proliferation was measured after 72 h of BRAFi treatment. a–c Proliferation of BRAF wt /KRAS G12D colorectal cancer cell lines after treatment with vemurafenib or PLX8394 at the indicated concentrations. Relative cell numbers are normalized to DMSO-treated control and differences shown as %. The tinted area indicates increased proliferation after treatment with vemurafenib. The Western blot inlay demonstrates the amount of ERK1/2 phosphorylation relative to the DMSO control at the concentration of vemurafenib that resulted in the biggest increase in proliferation. Lines between lanes denote non-adjacent lanes from the same membrane. d–e Inhibition of proliferation in BRAF V600E / KRAS wt colorectal cancer cell lines LIM2405 and COLO 201 after treatment with the indicated concentrations of vemurafenib or PLX8394. All data are shown as mean ± SD of independent triplicates relative to DMSO-treated controls
Conversely, BRAF V600E colon cancer cell lines LIM2405 and COLO 201 showed a decrease in pMEK and pERK levels with treatment (Fig. 1b, d, and f), consistent with reduced proliferation with both inhibitors (Fig. 2d and e).
Conclusions
We demonstrate that paradoxical activation of MAPK signalling and the consequent promotion of proliferation of KRAS-mutated colon cancer cells is markedly reduced by newer-generation paradox-breaker BRAF inhibitors, while their capacity to inhibit mutant BRAF-driven signalling is not compromised. Mechanistically, it has been demonstrated that this may be a consequence of the minor structural changes between paradox breakers and vemurafenib which most likely avoid paradoxical activation of MAPK signalling by preventing RAF dimer formation [16].
Amaravadi et al. reported that long-term BRAFi treatment can enhance neoplastic growth in colonic cells harbouring mutations of the tumour suppressor gene, adenomatous polyposis coli (APC), even without KRAS mutations [17]. While patient numbers in this study were small, 4 out of the 14 patients treated with conventional BRAFi presented with 5 or more colonic polyps, significantly increasing the potential risk for progression to colon cancer [18, 19]. Furthermore, using the APC Min +/− model, the authors demonstrated an increased number and shorter time to appearance of polyps in mice treated with vemurafenib compared with control. Collectively, these data emphasize the risks associated with long-term BRAFi treatment, and the applicability of these risks even to those patients with no prior history of RAS mutated cancer.
To sustain inhibition of MAPK signalling and to overcome paradoxical MAPK activation, BRAF inhibitors have been tested in combination with MEK inhibitors. Whilst this combination has been shown to improve survival times of patients with BRAF mutated melanoma [20], it is noteworthy that two of the cases of occult RAS mutated tumour progression on BRAFi therapy received, at least at some time-point, the BRAFi/MEKi combination [8, 11]. This suggests that the addition of a MEK inhibitor cannot completely abrogate BRAFi induced paradoxical MAPK activation. The mechanism of the paradox breaker’s ability to avoid paradoxical activation of the MAPK pathway has been previously demonstrated by Zhang et al. [16], using multiple cell lines including HCT 116. While the study showed similar paradoxical activation with vemurafenib in this cell line as demonstrated here, we extended these findings to additional colorectal carcinoma cell lines with different mutational backgrounds. Moreover, our study shows functional consequences of the paradoxical activation for the growth rate of the colorectal cancer cell lines, with a slight but consistent increase detected with vemurafenib treatment.
Overall, our findings justify the evaluation of paradox breaker BRAF inhibitors as the next generation of therapeutics for the treatment of BRAF mutated cancers. It suggests that the new paradox breakers have the potential to mitigate the risk of promoting occult RAS activated tumour progression associated with the use of the first generation BRAFi, without compromising therapeutic efficacy or narrowing the therapeutic window. Currently, PLX8394 is undergoing a clinical trial in solid unresectable tumors (NCT02428712) with a completion date of 2018, at which time point the usefulness of these drugs in the clinical setting will become clear.
References
Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KS. Paradoxical oncogenesis--the long-term effects of BRAF inhibition in melanoma. Nature Rev Clinical Onc. 2013;10:390–9.
Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, et al. PLX4032, a selective BRAF(V600E) kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAF melanoma cells. Pigment Cell Melanoma Res. 2010;23:190–200.
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5.
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30.
Oberholzer PA, Kee D, Dziunycz P, Sucker A, Kamsukom N, Jones R, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316–21.
Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–15.
Zimmer L, Hillen U, Livingstone E, Lacouture ME, Busam K, Carvajal RD, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375–83.
Carlino MS, Kwan V, Miller DK, Saunders CA, Yip D, Nagrial AM, et al. New RAS-mutant pancreatic adenocarcinoma with combined BRAF and MEK inhibition for metastatic melanoma. J Clin Oncol. 2015;33:e52–6.
Grey A, Cooper A, McNeil C, O'Toole S, Thompson J, Grimison P. Progression of KRAS mutant pancreatic adenocarcinoma during vemurafenib treatment in a patient with metastatic melanoma. Internal Med J. 2014;44:597–600.
Callahan MK, Rampal R, Harding JJ, Klimek VM, Chung YR, Merghoub T, et al. Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367:2316–21.
Andrews MC, Behren A, Chionh F, Mariadason J, Vella LJ, Do H, et al. BRAF inhibitor-driven tumor proliferation in a KRAS-mutated Colon carcinoma is not overcome by MEK1/2 inhibition. J Clin Oncol. 2013;31:e448–51.
Gayet J, Zhou XP, Duval A, Rolland S, Hoang JM, Cottu P, et al. Extensive characterization of genetic alterations in a series of human colorectal cancer cell lines. Oncogene. 2001;20:5025–32.
Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100.
Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014;27:479–84.
Choi J, Landrette SF, Wang T, Evans P, Bacchiocchi A, Bjornson R, et al. Identification of PLX4032-resistance mechanisms and implications for novel RAF inhibitors. Pigment Cell Melanoma Res. 2014;27:253–62.
Zhang C, Spevak W, Zhang Y, Burton EA, Ma Y, Habets G, et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature. 2015;526:583–6.
Amaravadi RK, Hamilton KE, Ma X, Piao S, Portillo AD, Nathanson KL, et al. Multiple gastrointestinal polyps in patients treated with BRAF inhibitors. Clin Can Res. 2015;21:5215–21.
Schuman BM, Simsek H, Lyons RC. The association of multiple colonic adenomatous polyps with cancer of the colon. Am J Gastroenterology. 1990;85:846–9.
Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, et al. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res. 2014;74:3238–47.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703.
Suardet L, Gaide AC, Calmes JM, Sordat B, Givel JC, Eliason JF, et al. Responsiveness of three newly established human colorectal cancer cell lines to transforming growth factors beta 1 and beta 2. Cancer Res. 1992;52:3705–12.
Whitehead RH, Zhang HH, Hayward IP. Retention of tissue-specific phenotype in a panel of colon carcinoma cell lines: relationship to clinical correlates. Immunol Cell Biol. 1992;70(Pt 4):227–36.
Semple TU, Quinn LA, Woods LK, Moore GE. Tumor and lymphoid cell lines from a patient with carcinoma of the colon for a cytotoxicity model. Cancer Res. 1978;38:1345–55.
Acknowledgements
We thank Plexxikon for the supply of BRAF inhibitors PLX8394 and PLX7904, and Dr. Liliana Endo-Munoz for input in the preparation of this manuscript.
Funding
This research was supported by a Melanoma Research Alliance Team Science Award to JC and in part by Operational Infrastructure Support Program Funding of the Victorian State Government for the Ludwig Institute for Cancer Research. AB is supported by a Cancer Council Victoria grant, MCA is supported by a National Health & Medical Research Council of Australia Postgraduate Research Scholarship (NHMRC#1055456), and JMM is supported by a NHMRC Research Fellowship (APP1046092).
Availability of data and materials
All data generated during this study are included in this published article and its additional information files.
Author’s contributions
CSAT, MCA, PI, CH and AB derived the cell lines, performed the protein extractions, western blots and cellular proliferation assays. CSAT and AB were major contributors in writing the manuscript. JC, JM and AB made substantial contributions to the conception and design of the study, and the interpretation of data, and were involved in critically revising the manuscript for important intellectual content. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All research involving human participants or tissue carried out at the Olivia Newton-John Cancer Research Institute is conducted under the guidelines of the Human Research Ethics Committee (HREC) of the Austin Health Office for Research. The HREC conforms with the National Statement on Ethical Conduct in Human Research (NHMRC, ARC, UA, 2007, National Statement) and is constituted in accordance with the requirements of the National Health & Medical Research Council (NHMRC). Both committees operate under strict Terms of Reference and Standard Operating procedures.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Material and Methods. (DOCX 19 kb)
Additional file 2: Figure S1.
The effect of vemurafenib and PLX8394 on the BRAF V600E melanoma cell line LM-MEL-64 and the BRAF wt melanoma cell line LM-MEL-39. (A) LM-MEL-64 and (B) LM-MEL-39 were treated with the indicated concentrations of vemurafenib or PLX8394 and immunoblotting for total and phosphorylated ERK was performed. (C) pERK densitometry relative to control expressed as (%) ± SD for LM-MEL-64 and (D) for LM-MEL-39. Data are from three independent experiments. (TIFF 342 kb)
Additional file 3: Figure S2.
The effect of BRAF inhibitors vemurafenib and PLX8394 on BRAF wt / KRAS G13D cell line HCT 116. Cells were treated with DMSO, vemurafenib at 1 μM, or PLX8394 at 1 μM for 6 h. (A) Representative Western blot after treatment with DMSO control or BRAF inhibitors. Western blots were probed for total and phosphorylated MEK1/2 and ERK1/2. The blots are representative of three independent experiments. GAPDH served as a loading control. Western blot signal intensity was quantified and used to measure protein level relative to control. (B) Densitometry of MEK1/2 phosphorylation demonstrating paradoxical activation by vemurafenib in HCT 116. (C) Densitometry of ERK1/2 phosphorylation in the same cell line. Total protein:phosphorylated protein ratio is expressed as the mean ± SD of three independent replicates relative to DMSO-treated control. (D) Inhibitors were used at 0 (DMSO control), 0.1, 0.5, and 1 μM. Cell proliferation was measured after 72 h of BRAFi treatment. Relative cell numbers are normalized to DMSO-treated control and differences shown as percentage. The tinted area indicates increased proliferation after treatment with vemurafenib. The Western blot inlay demonstrates the difference in ERK1/2 phosphorylation at the concentration of vemurafenib that resulted in the biggest increase in proliferation. (TIFF 1052 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tutuka, C.S.A., Andrews, M.C., Mariadason, J.M. et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol Cancer 16, 112 (2017). https://doi.org/10.1186/s12943-017-0684-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-017-0684-x